Compare means from baredSC results¶
baredSC outputs can be used to evaluate the fold-change of expression between 2 conditions.
Inputs¶
We use the same inputs as in previous input descriptions.
Run baredSC on each subpopulation¶
Let’s focus on cells of group 0.0 and group 3.0 (they correspond to each of the 2 gaussians found previously).
To increase the speed we will use less bins (--nx 25):
$ for group in 0 3; do
for nnorm in 1 2; do
baredSC_1d \
--input example/nih3t3_generated_2d_2.txt \
--metadata1ColName 0.5_0_0_0.5_group \
--metadata1Values ${group}.0 \
--geneColName 0.5_0_0_0.5_x \
--output example/first_example_1d_group${group}_${nnorm}gauss_25_neff200 \
--nnorm ${nnorm} --nx 25 --minNeff 200 \
--figure example/first_example_1d_group${group}_${nnorm}gauss_25_neff200.png \
--title "first gene ${nnorm} gauss group${group} 25 bins neff200" \
--logevidence example/first_example_1d_group${group}_${nnorm}gauss_25_neff200_logevid.txt
done
combineMultipleModels_1d \
--input example/nih3t3_generated_2d_2.txt \
--metadata1ColName 0.5_0_0_0.5_group \
--metadata1Values ${group}.0 \
--geneColName 0.5_0_0_0.5_x --nx 25 \
--outputs example/first_example_1d_group${group}_1gauss_25_neff200 \
example/first_example_1d_group${group}_2gauss_25_neff200 \
--figure example/first_example_1d_group${group}_1-2gauss_25.png \
--title "first gene group$group 25 bins 1 and 2 gauss"
done
In one case, 100 000 samples were not sufficient.
We check the QC. We can now compare the results:
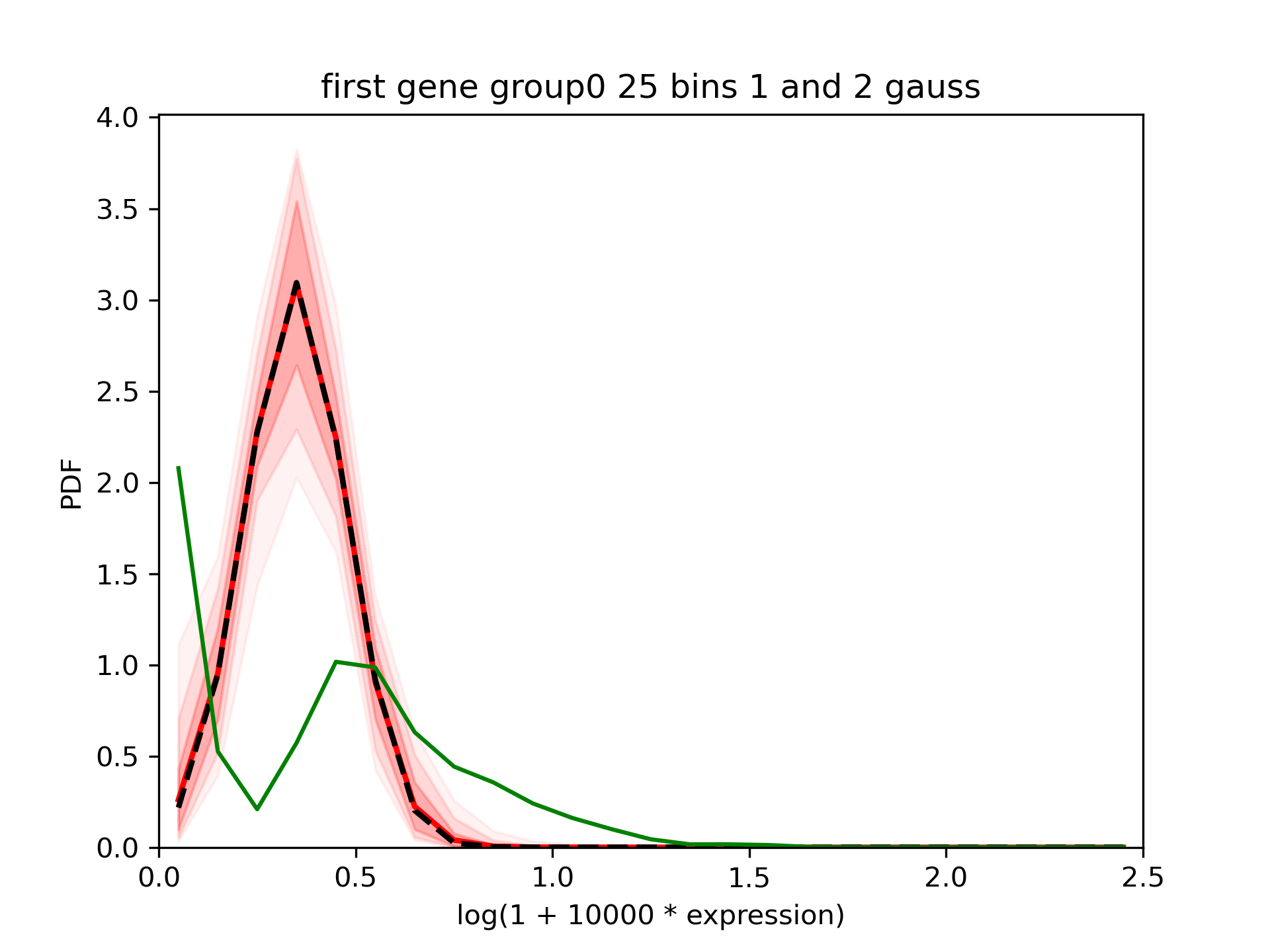
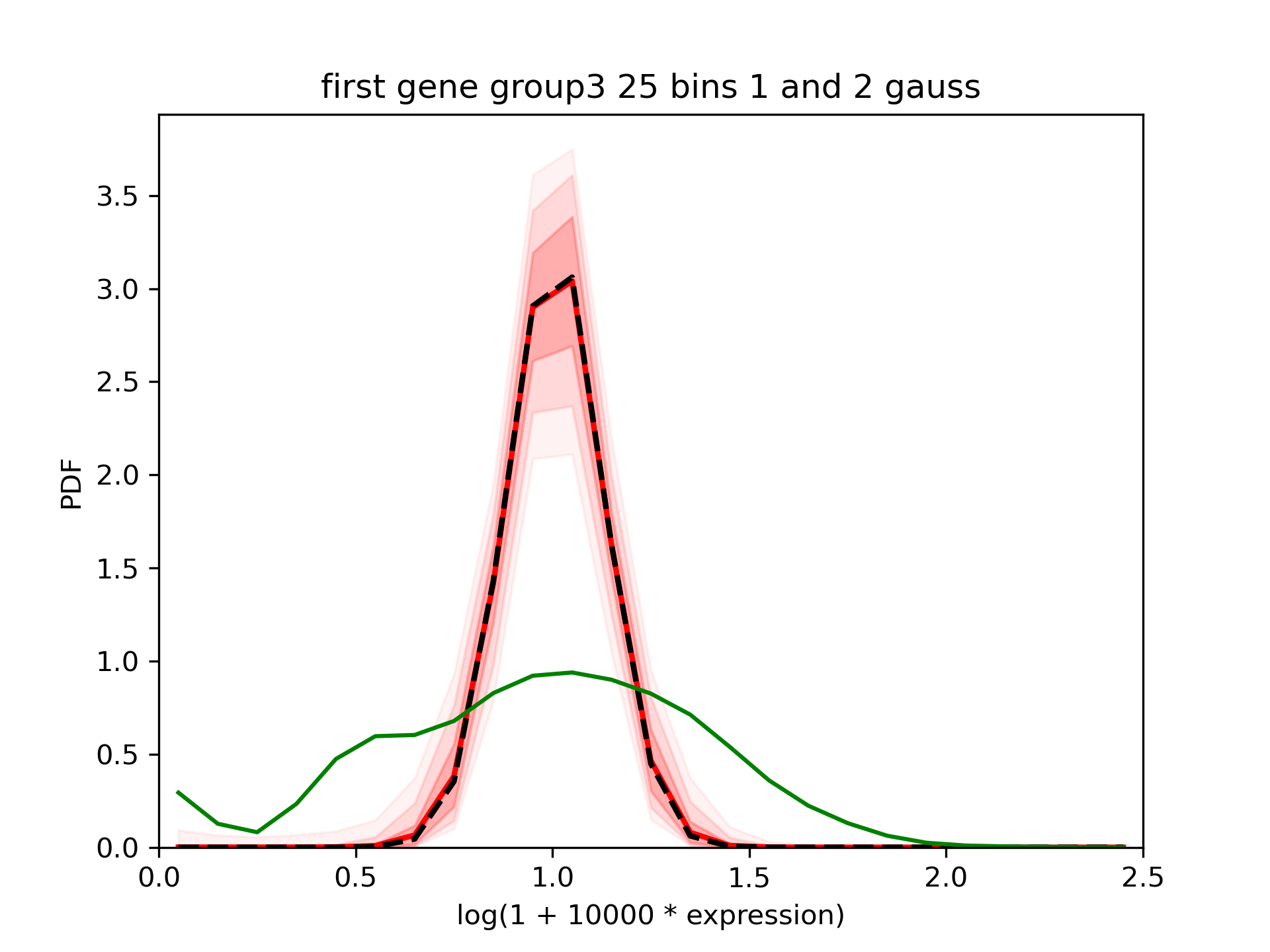
We can see that the tool fits relatively nicely the gaussians which were in inputs.
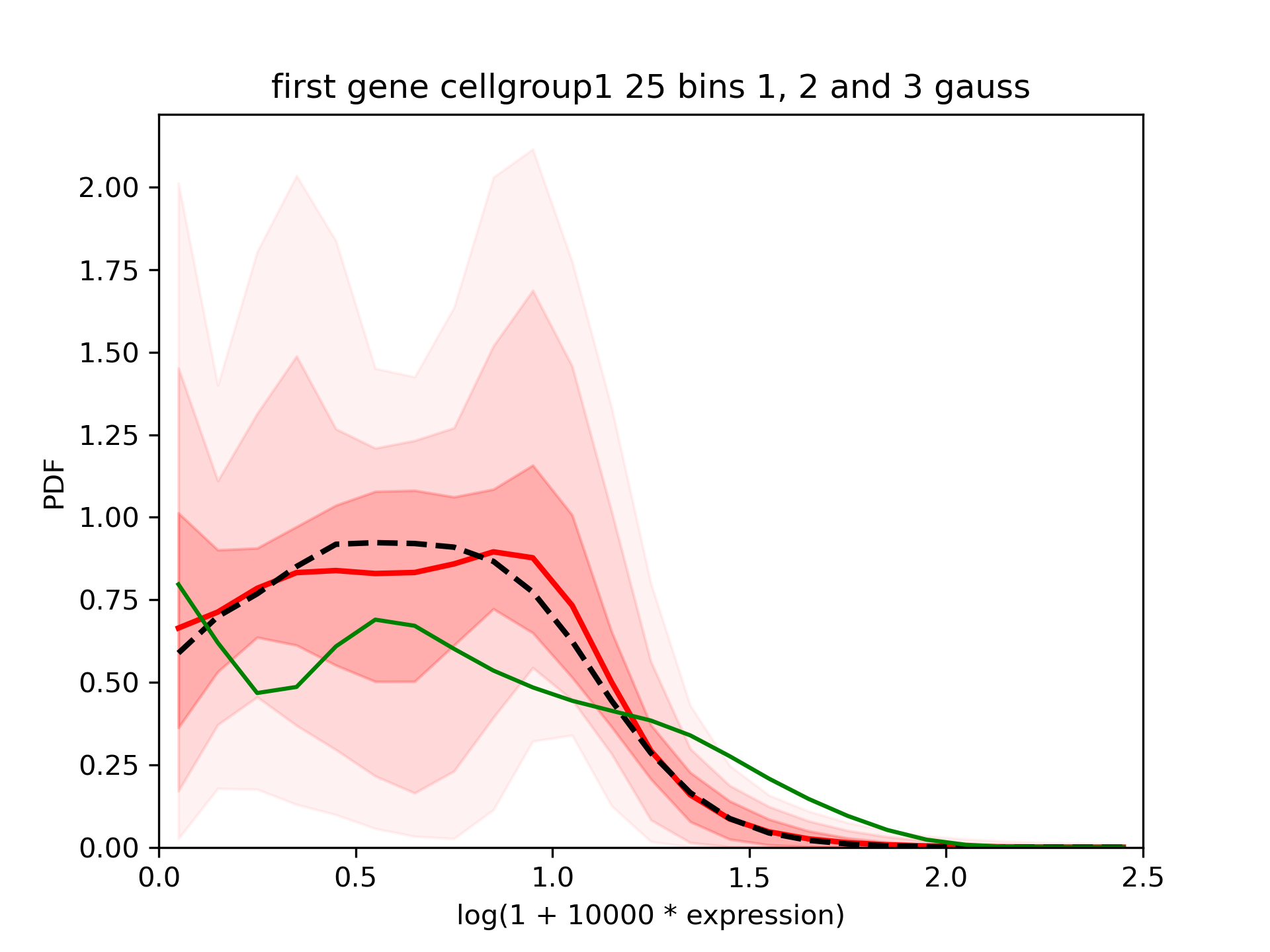
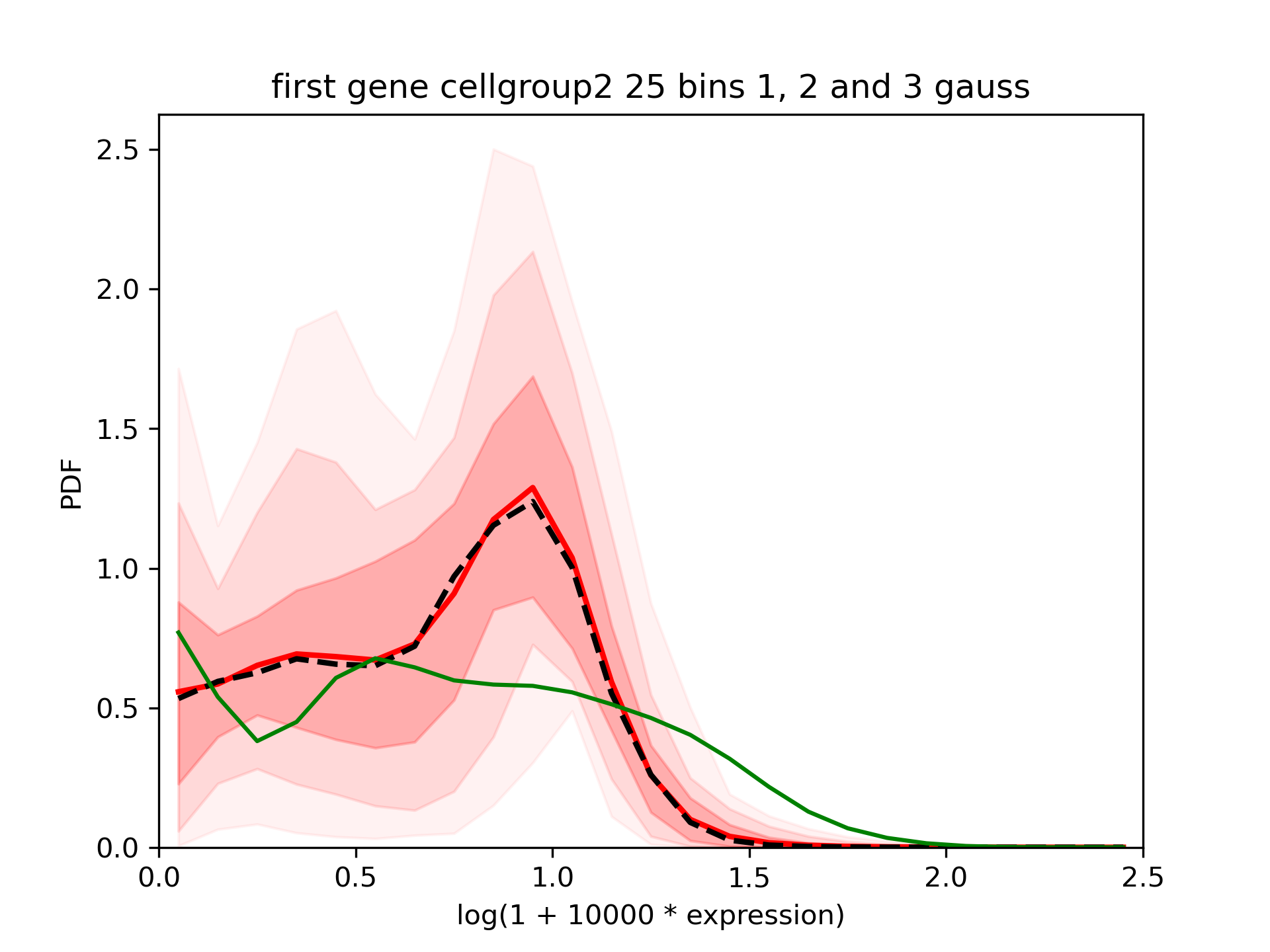
If we use another metadata which is just 300 and 500 random cells:
$ for group in 1 2; do
for nnorm in 1 2 3; do
baredSC_1d \
--input example/nih3t3_generated_2d_2.txt \
--metadata1ColName group \
--metadata1Values group${group} \
--geneColName 0.5_0_0_0.5_x \
--output example/first_example_1d_cellgroup${group}_${nnorm}gauss_25_neff200 \
--nnorm ${nnorm} --nx 25 --minNeff 200 \
--figure example/first_example_1d_cellgroup${group}_${nnorm}gauss_25_neff200.png \
--title "first gene ${nnorm} gauss cellgroup${group}" \
--logevidence example/first_example_1d_cellgroup${group}_${nnorm}gauss_25_neff200_logevid.txt
done
combineMultipleModels_1d \
--input example/nih3t3_generated_2d_2.txt \
--metadata1ColName group \
--metadata1Values group${group} \
--geneColName 0.5_0_0_0.5_x --nx 25 \
--outputs example/first_example_1d_cellgroup${group}_1gauss_25_neff200 \
example/first_example_1d_cellgroup${group}_2gauss_25_neff200 \
example/first_example_1d_cellgroup${group}_3gauss_25_neff200 \
--figure example/first_example_1d_cellgroup${group}_1-3gauss_25.png \
--title "first gene cellgroup$group 25 bins 1, 2 and 3 gauss"
done
We see that the results are different in both groups but in both cases the confidence interval is quite large:
One of the output is *_means.txt.gz. Each line correspond to the value of the mean expression evaluated at each sample of the MCMC. It can be used to estimate:
A confidence interval on the mean value (in the used axis log(1 + 10^4 * expression))
A confidence interval on the fold change (delogged).
In [1]: import numpy as np
...: def delog(x):
...: return(1e-4 * (np.exp(x) - 1))
...: my_quantiles = [0.5 - 0.6827 / 2, 0.5, 0.5 + 0.6827 / 2]
...: # Get data
...: means1 = np.genfromtxt('../example/first_example_1d_group0_1-2gauss_25_means.txt.gz')
...: print(f'Values of mean in group0: {means1}')
...: means2 = np.genfromtxt('../example/first_example_1d_group3_1-2gauss_25_means.txt.gz')
...: print(f'Values of mean in group3: {means2}')
...: # Shuffle means2
...: np.random.shuffle(means2)
...: print(f'Mean log expression in group0: {np.quantile(means1, my_quantiles)}')
...: print(f'Mean log expression in group3: {np.quantile(means2, my_quantiles)}')
...: fc = [delog(x1) / delog(x2) for x1, x2 in zip(means1, means2)]
...: print(f'Estimation of fold-change: {np.quantile(fc, my_quantiles)}')
...:
Values of mean in group0: [0.35776993 0.35776993 0.35776993 ... 0.34672964 0.34672964 0.38877004]
Values of mean in group3: [1.00746002 1.02085388 1.02085388 ... 1.00369734 1.01363153 1.02256951]
Mean log expression in group0: [0.33838793 0.34953488 0.36060358]
Mean log expression in group3: [0.99479228 1.00628287 1.01751424]
Estimation of fold-change: [0.23110761 0.24112418 0.25130651]
We can use matplotlib to display the results graphically:
In [2]: import matplotlib.pyplot as plt
...: x1, x2 = np.random.normal(1, 0.1, len(means1)), np.random.normal(3, 0.1, len(means2))
...: fig, axs = plt.subplots(1, 2)
...: axs[0].scatter(x1, means1, s=1, alpha=0.01)
...: axs[0].scatter(x2, means2, s=1, alpha=0.01)
...: axs[0].set_ylim(0, )
...: axs[0].set_ylabel('log(1 + expression * 10\'000)')
...: axs[0].set_xticks([1, 3])
...: axs[0].set_xticklabels(['group0', 'group3'], fontsize='x-large')
...: axs[1].scatter(x1[:len(fc)], fc, s=1, alpha=0.01)
...: axs[1].set_xticks([1])
...: axs[1].set_xticklabels(['fold-change'], fontsize='x-large')
...: axs[1].set_ylim(0, )
...: axs[1].set_ylabel('Fold-change (not log transformed)')
...: fig.tight_layout()
...:
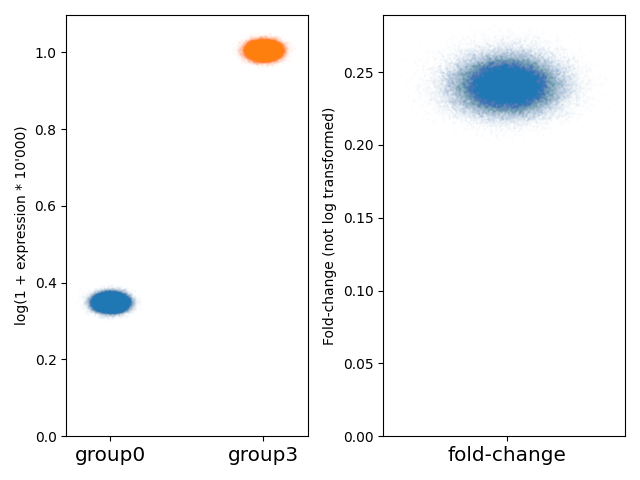
On the first example with group0 and group3 where each is a different gaussian, we see a mean log expression around the expected 0.375 value in group 0, 1 in group 3. We see that the fold-change is around 25% (this is the real fold-change, not log transformed).
Now if we have a look to the subsamples of cells (300 and 500 cells) and perform the same analysis:
In [3]: # Get data
...: means1 = np.genfromtxt('../example/first_example_1d_cellgroup1_1-3gauss_25_means.txt.gz')
...: print(f'Values of mean in cellgroup1: {means1}')
...: means2 = np.genfromtxt('../example/first_example_1d_cellgroup2_1-3gauss_25_means.txt.gz')
...: print(f'Values of mean in cellgroup2: {means2}')
...: # Shuffle means2
...: np.random.shuffle(means2)
...: print(f'Mean log expression in cellgroup1: {np.quantile(means1, my_quantiles)}')
...: print(f'Mean log expression in cellgroup2: {np.quantile(means2, my_quantiles)}')
...: fc = [delog(x1) / delog(x2) for x1, x2 in zip(means1, means2)]
...: print(f'Estimation of fold-change: {np.quantile(fc, my_quantiles)}')
...:
Values of mean in cellgroup1: [0.66035127 0.66035127 0.62547552 ... 0.62783495 0.62783495 0.62783495]
Values of mean in cellgroup2: [0.68203872 0.69214061 0.71198453 ... 0.70698152 0.70698152 0.70698152]
Mean log expression in cellgroup1: [0.6161945 0.64620379 0.67630369]
Mean log expression in cellgroup2: [0.66763529 0.69110072 0.71426981]
Estimation of fold-change: [0.8423754 0.91214824 0.9865969 ]
In [4]: x1, x2 = np.random.normal(1, 0.1, len(means1)), np.random.normal(3, 0.1, len(means2))
...: fig, axs = plt.subplots(1, 2)
...: axs[0].scatter(x1, means1, s=1, alpha=0.01)
...: axs[0].scatter(x2, means2, s=1, alpha=0.01)
...: axs[0].set_ylim(0, )
...: axs[0].set_ylabel('log(1 + expression * 10\'000)')
...: axs[0].set_xticks([1, 3])
...: axs[0].set_xticklabels(['cellgroup1', 'cellgroup2'], fontsize='x-large')
...: axs[1].scatter(x1[:len(fc)], fc, s=1, alpha=0.01)
...: axs[1].set_xticks([1])
...: axs[1].set_xticklabels(['fold-change'], fontsize='x-large')
...: axs[1].set_ylim(0, )
...: axs[1].set_ylabel('Fold-change (not log transformed)')
...: fig.tight_layout()
...:
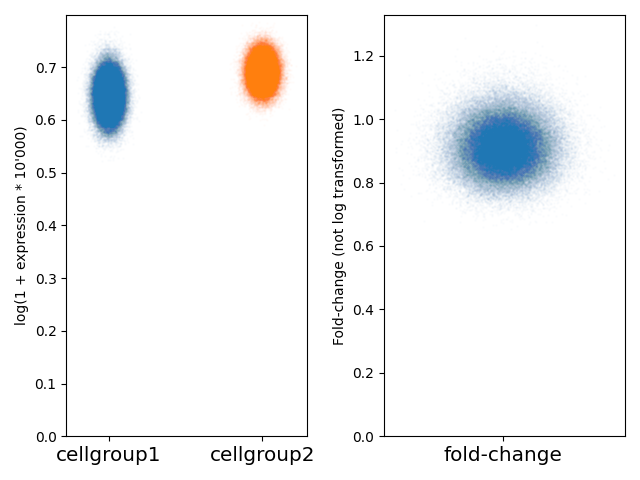
Here, we can see that the fold-change is slightly below 1.